We are interested in understanding how the demography of wild and domesticated populations can impact patterns of genetic variation. In particular, we focus on understanding how small populations can accumulate harmful (“deleterious”) mutations due to
We are interested in understanding how the demography of wild and domesticated populations can impact patterns of genetic variation. In particular, we focus on understanding how small populations can accumulate harmful (“deleterious”) mutations due to increased genetic drift and inbreeding, and how these processes can lead to fitness reductions and potentially extinction. Numerous recent and ongoing projects in the lab have investigated the dynamics of deleterious mutations in small populations, focusing on a variety of species including breed dogs, the Channel Island fox, sea otters, caracals, and the wolves and moose of Isle Royale, Michigan. These studies combine empirical genome sequencing data with simulations to detail the role that population size plays in determining the fate of deleterious mutations. Several of our studies have highlighted the importance of purging (purifying selection against recessive deleterious mutations) as a mechanism that enables the long-term persistence of small populations. These projects have been conducted in collaboration with the Lohmueller Lab.
Notable recent papers on these topics include:
Marsden et al. 2016 which found an increased burden of deleterious variation in dogs due to the domestication bottleneck.
Robinson et al. 2016 and 2018 which determined that island foxes on the Channel Islands of California have a complicated history of population bottlenecks that has led to virtually no genetic diversity, coining the term “genomic flatlining.” The foxes appear to have persisted in spite of their extreme lack of diversity by purging highly deleterious variants.
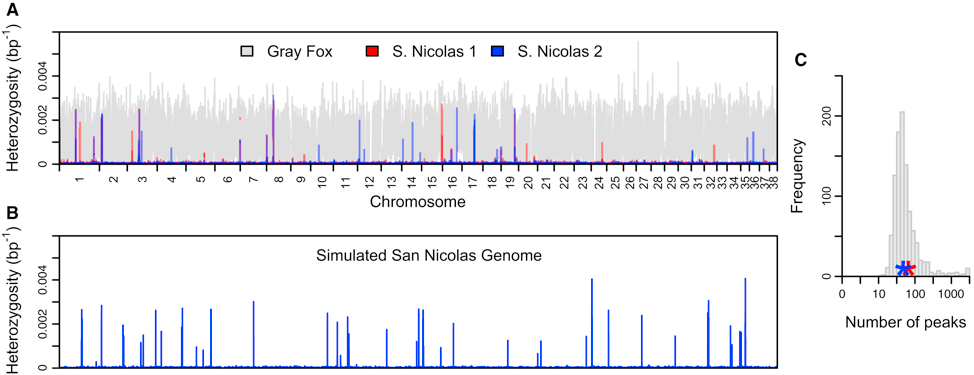
Figure from Robinson et al. 2016. Genome wide heterozygosity (in 1-kb sliding windows) of Channel Island foxes compared to mainland gray foxes.
Beichman, Phung and Lohmueller (2017) demonstrated that commonly used demographic techniques in conservation genetics can be dangerously misleading if taken as literal records of population history. Beichman, Huerta-Sanchez and Lohmueller (2018) presented a review paper for those working on non-model organisms, especially conservation geneticists, to carry out demographic inference from genomic data.
Robinson et al. (2019) investigated the genomic consequences of severe inbreeding depression in the isolated wolf population of Isle Royale, Michigan.
Beichman et al. (2019) carried out comparative genomic analyses on the sea otter and giant otter genomes, finding a lack of diversity in the sea otter genome that reflects long-term low population size for the species.
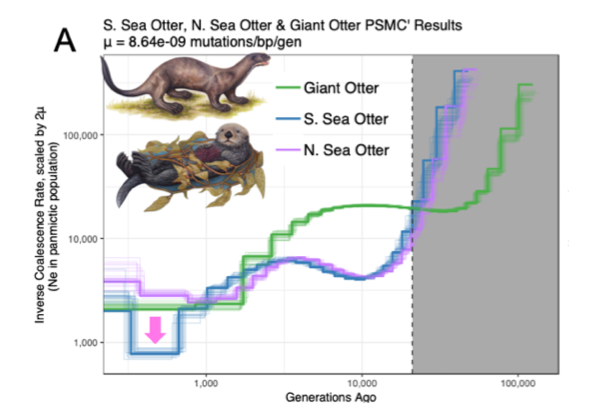
Figure from Beichman et al. 2019. Whole-genome based demographic inference of southern and northern sea otters and the South American giant otter.
Beichman et al. (in preparation) uses whole-exome and neutral region sequencing of five remnant sea otter populations to determine patterns of population structure and infer the impact of the fur trade bottleneck.
Kyriazis et al. (in revision) used ecologically-realistic population genetic simulations to investigate the role that demography and strongly deleterious mutations play in determining extinction risk in small populations.